
Clinical correlation of Process 2 lots
Regulators knew and approved the untrialed jabs on adults, pregnant women, children and babies

Fom Josh Guetzkow https://x.com/joshg99/status/1658421192326365185?s=20
Was the Pfizer/BioNTech vaccine clinical trial a bait-and-switch? There were >44,000 people in the trial, but only ~250 of them were given doses made with a new manufacturing method ('process 2') that was used to make enough doses to sell around the world.
The pivotal clinical trial protocol amendment
EMA EPAR Comirnaty see page 69/140
The European Public Assessment Report is the official initial assessment of the vaccine by the European Medicines Agency, the regulator for the EU.

As you can see, 250 patients were supposed to get Process 2 jabs to compare to Process 1 jabs to ensure that clinical outcomes would be similar to that seen in the clinical trial
In the supplement to the pivotal trial published in the NEJM by Polack et al, is the protocol amendment. See page 2/3 of Final Protocol starting at Page 128. Supplementary Appendix to Polock et al Protocol Amendment 7 (Oct 06, 2020) and Protocol Amendement 9 (October 9, 2020) that they would be selected randomly.
“...each lot of Process 2-manufactured BNT162b2 will be administered to approximately 250 participants 16 to 55 years of age. The safety and immunogenicity of prophylactic BNT162b2 in individuals 16-55 years of age vaccinated with ‘Process 1” and each lot of “process 2” study intervention will be described. A random sample of 250 participants from those vaccinated with study intervention produced by manufacturing “Process 1” will be selected for this descriptive analysis.”
Josh Guetzkow in his May 13, 2023 letter to the BMJ Letter to BMJ tries to determine the outcome of this protocol amendment.
Two documents obtained through a Freedom of Information Act (FOIA) request[6] describe the vaccine batches and lots supplied to each of the trial sites through November 19, 2020[7] and March 17, 2021,[8] respectively. According to these documents, doses from ‘Process 2’ batch EE8493Z are listed at four trial sites prior to November 19, and four other sites are listed with ‘Process 2’ batch EJ0553Z in the updated document. Both batches were also part of the emergency supply for public distribution. The CDC’s Vaccine Adverse Event Reporting System, known to be underreported,[9] lists 658 reports (169 serious, 2 deaths) for lot EE8493[10] and 491 reports (138 serious, 21 deaths) for lot EJ0553.[11]
He also notes:
The 6-month interim clinical study report[12] from the Comirnaty trial notes that “the IR for any AE and at least 1 related AE and severe AE for participants who originally received placebo and then received BNT162b2 are greater (205.4 per 100 PY, 189.5 per 100 PY, 6.0 per 100 PY) than the IRs (83.2 per 100 PY, 62.9 per 100 PY, 4.3 per 100 PY) for participants who originally were randomized to BNT162b2” (p222). It is unclear whether there is a connection between the lots administered to the crossover placebo subjects and the elevated rate of AE’s.
Were these patients, originally who received placebo, given Process 2 batches which resulted in a higher adverse event rate? It is not clear if these crossover patients received Process 2 batches since Guetzkow notes
additional ‘Process 1’ batch EE3813 doses with distinct Pfizer lot numbers were added to the later batch document[7] at over 70% of trial sites, potentially supplied at a later stage to enable vaccination of placebo patients with BNT162b2.
Panic at the EMA
The clinical correlation between Process 1 and Process 2 jabs was a concern with the regulators primarily due to the loss of RNA integrity. This would imply that the efficacy of the Process 2 jabs would be less since it is the intact modRNA which is the template for spike protein or antigen production. Less modRNA would presumably result in less spike protein and therefore less antibodies and decreased efficacy.
This is summed up in an email from an the Head of Pharmaceutical Quality on NOVEMBER 24, 2020 (from EMA leak/hack)

Note PPQ=process performance lots. These would be the standard lots with quality criteria to which all subsequent lots are compared. DS=drug substance of the modRNA and DP=final drug product ready to be dispensed (ie modRNA/LNPs in final formulation)
Shortly thereafter there is a telephone conference with the FDA and Health Canada on this issue, dated NOVEMBER 25, 2020.

Here we see the mention of the clinical trial (CT) protocol amendment insisted upon by the FDA to determine the clinical correlation between Process 1 and Process 2. In addition, specifications are to be agreed upon jointly. Will an RNA integrity value of 70% be agreed upon? It appears not, since it ended up being 55% at the time it was approved. The truncated and fragmented mRNA (variable species) that were identified as a safety risk by the EMA, was considered less likely by the FDA and Health Canada. This assumption may be premature, however. Additionally, we now have ANOTHER serious issue regarding the formulation and visible particles. Visible particles in a sterile parenteral solution (IM or IV) is associated with adverse events (especially a nanoparticle formulation) and a sign of poor formulation. Formulation is the buffered, sucrose solution in which the LNPs are dispersed.
Regulatory Guidelines for Process Changes During Manufacturing
Manufacturers frequently make changes to the manufacturing process pre and post approval as more information is gained and as the processes improve. As a result there are guidelines to determine the level and amount of data needed to evaluate if the new manufacturing process results in an equivalent drug. For small organic molecules like drugs, one would expect a very close comparison (often 97% or higher). For biologics like vaccines or monoclonal antibodies, it is essentially impossible to obtain the exact same molecule as the active ingredient, antigen or antibody since the product isn’t a pure single amino acid sequence to begin with. There may be a little variation. For comparability between biological products, it is expected that
“the manufacturing process changes will not have an adverse impact on the quality, safety and efficacy of the drug product.”
see International Council For Harmonization Q5E
Under this guidance, the requirements for clinical comparative efficacy and safety studies are dependent on the stage of development and the type of change involved. If changes are made AFTER the confirmatory clinical trials, as was done with the Pfizer/BioNTech pro-vaccine, a thorough comparability assessment is generally required including:
“…physicochemical and biological in vitro studies, and may include clinical pharmacokinetic and/or pharmacodynamic comparability studies. If this comparability exercise cannot rule out an impact on the efficacy and safety profile of the drug, additional clinical study (ies) may have to be performed.” (pg 6/10)
Well since the side to side comparability exercise between Process 1 and Process 2 includes a drop in the drug substance and now visible particles in the final formulation, some kind of clinical study (its) may have to be performed.
What can be done? Pressure on the EMA.
Earlier, there was pressure from the Commissioner of the EU (Ursula van de Leyden) that appeared quite tense. Insistence that the vaccines be approved regardless of issues of manufacturing included using threats of invoking Article 5(2) of the EU.

Days before the teleconference with the EMA, on November 23, 2020, Alexis Nolte (Head of the Human Medicines Division of the EMA) suggests an animal or other test to determine if protein expression is affected, assuming amount of mRNA is proportional to amount of spike protein produced. However, data on clinical efficacy can ONLY be assured with a clinical trial in humans, the protocol amendment to the clinical trial which was still ongoing when this email was sent.

Correlation of intact modRNA and antibody response. Is this the “animal” correlate?
Hidden away in an Annex from the EMA leak is an FDA document which has a small number of mice being inoculated with what I presume are Process 2 lots.
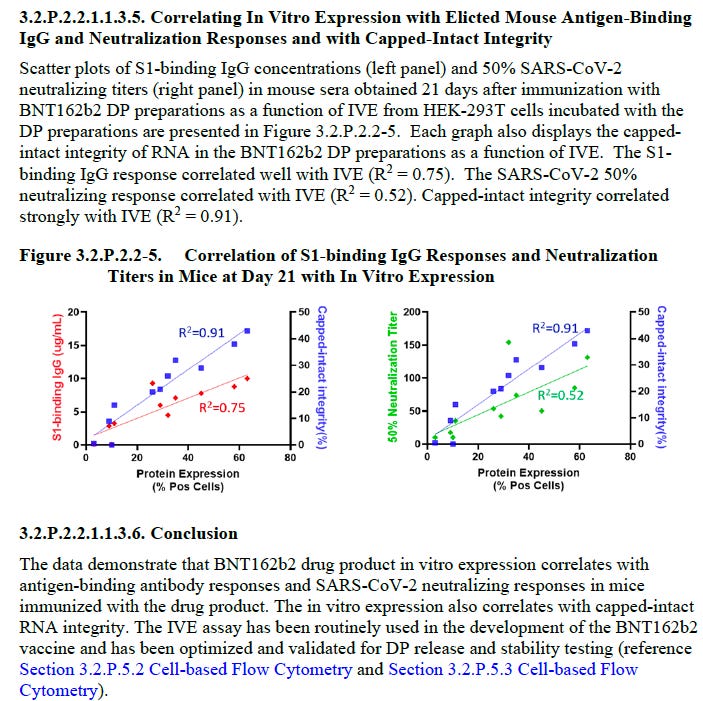
So fully capped mRNA is required to exert and antibody response from spike protein expression. There is a correlation of 0.52 with neutralizing antibodies and amount of capped mRNA in the jab, but the in vitro expression (IVE) showed a 0.91 correlation. I think this means not all spike protein produced will elicit antibodies but that is probably normal (??).
Note we don’t know how many mice, nor the test item (batch number) or any other detail. More digging is required.
Meeting between Pfizer and the EMA
Ongoing issues of the comparability between Process 1 and Process 2 was discussed at an internal meeting EMA had with its advisory panel. Questions had been posed to Pfizer/BioNTech and written answers obtained. Here the EMA is stating the clinical correlation which was included in the pivotal trial has now been excluded since the interim analysis was changed and that data wont be available (likely until the new year). Pfizer/BioNTechs response is that they are good. The specifications are good and therefore the clinical correlation in actual humans were not required.
Covid-19 Rapporteur/CMC Meeting November 26, 2020
This was after the teleconference with the FDA and HC. (from EMA leak/hack)

As Geoff Pain states, Pfizer/BioNTech HAD NO INTENTION of doing a clinical correlate from the very beginning.
EMA Approves BNT162b on Dec 21, 2020
The EMA approves the Pfizer/BioNTech vaccine on Dec 21, 2020 under Conditional Marketing Approval (CMA) that was subjected to several Specific Obligations.However, none of these Specific Obligations including data on the clinical correlation in patients but did require more extensive testing and and safety considerations on the manufacturing process. At the time of the CMA, it does not appear that data on the clinical correlation between Process 1 and Process 2 lots were available. Therefore there was no verification that the results of the clinical trials using Process 1 would translate into the same efficacy and safety with the Process 2 lots. Did Public Health officials know? Were patients told?
As mentioned earlier, emails stated if the EMA did not approve the vaccine by the end of December then Article 5(2) would be triggered by the Commissioner. This means if there are divergent opinions for authorization by the member states or if the EMA recommends a delay or refuses authorization, the Commissioner can just overturn or ignore the EMA recommendation and authorize the vaccine for all member states. No wonder the EMA wanted to avoid that scenario.
What was the outcome? Did the EMA/FDA get the promised data?
Verification with FOIA answers from MHRA (UK) as provided by Nick Hunt in the Daily Skeptic
Here's the relevant extract from the MHRA FOI reply :
2. Pfizer amended C4591001 in October 2020 to add, inter alia, at para
9.4 : "The safety and immunogenicity results for individuals 16 to 55
years of age vaccinated with study intervention produced by
manufacturing “Process 1” and each lot of “Process 2” will be
summarized descriptively. A random sample of 250 participants from
those vaccinated with study intervention produced by manufacturing
“Process 1” will be selected randomly for the analysis." Request 2:
Please can you send me a copy of Pfizer's report of this.
Alternatively, if you exempt its release, tell me the Pfizer reference
and date.
Answer:
Under section 1(1)(a), the review confirms a report specifically on
analysis on the “random sample of 250 participants vaccinated with
study intervention produced by manufacturing “Process 1”” is not held.
To provide helpful context and background, in the early stages of the
pandemic, before BNT162b2 was authorised or approved, improvements
were made to the manufacturing process to adjust the scalability,
robustness, and productivity in preparation for large scale
manufacture (Process 2); scaling of manufacturing processes is a
common occurrence in the manufacture of medicines. Manufacturing steps
that were not scalable were replaced with those designed to provide a
similar or better impurity profile. This “process 2 drug” substance
was shown to be comparable through side-by-side comparability studies
and heightened characterisation testing. The process was validated at
all manufacturing sites and submitted for review and approval.
Vaccines produced by both “Process 1” and “Process 2” were included in
the pivotal clinical trial (C4591001). Typically, such changes can be
supported by analytical data; however, due to the nascent regulatory
landscape for COVID-19 vaccines, in October 2020 an exploratory
objective was added in the C4591001 study to describe safety and
immunogenicity of vaccines produced by manufacturing “Process 1” or
“Process 2” in participants 16 to 55 years of age. This exploratory
objective was removed and documented in protocol amendment 20 in
September 2022 due to the extensive usage of vaccines manufactured via
“Process 2”. Thus, this process comparison was not conducted as part
of the formal documentation within the protocol amendment.
Best regards,
Nick
Did you get that? Clinical correlation between the two processes are no longer required since, well, we have real world evidence that supports the efficacy and safety of Process 2.
That protocol amendment never done. WHY? It appears there was NO INTENT to actually determine if the new vaccine was comparable to the vaccine used in the trial. Because of the change in starting materials, purification processes and analytical tests used for Process 2 lots, as well as differences in the actual amount of “active substance” in the vaccines, it CANNOT be assumed the result will be comparable. There is not enough knowledge from this new platform to waive this requirement.
Coincidently, just recently, NEW REGULATORY GUIDELINES USING REAL WORLD EVIDENCE FROM THE FDA has been released. Normally, protocol amendments that are not completed would have resulted in a suspension of the vaccine, or limited use in selected populations as these differences in a biological product would normally result in suspension of regulatory approval until fixed. Will these new guidelines be used to approve the Process 2 lots as comparable to the Process 1 lots?
FDA Guidelines on Real World Data/Evidence
IMPLICATIONS
the commercial vaccine lots were truly experimental without even a basis of comparison to the clinical trial
there was and still is no informed consent
these vaccines were used in babies, the immunocompromised, pregnant women etc without any assurances that these would even be EFFECTIVE, let alone safe
Further regulatory approvals for the booster, the bivalents and the new Omicron XBB vaccine are based on a MIRAGE since we cannot anchor the commercial lots to the clinical trial. Do you know what that means? All the approvals are bogus on legal regulatory grounds, irrespective if they have 8 mice or not. Dig deep and there is no foundation on science or evidence or on normal processes for these process 2 lots. Although many think the FDA is captured, and it is, most of the written process and procedures are followed by both sides. It benefits both parties.
Besides the regulators, the manufacturers are not providing data and processes when required or asked. This is not done. The FDA/EMA wants this, you give it to the best of your ability. Otherwise you cannot sell your drug and sell your drug is what Pharma wants. Therefore, they knew they would get approved regardless, did not need to follow GMP, or clinical trial guidelines, did not need to provide requested data and did not need to provide safe labels or product information to health care providers. Lest you think this is normal, it is not. Pharma is very proud of their manufacturing prowess and that is one aspect that Pharma rarely cuts corners, at least for new innovative drugs. They do this because it is harder for a generic or say a manufacturer in India to copy. I have to say, what are the other manufacturers thinking? Will they also get special treatment? If not, why not?
Real world evidence was not used for this kind of assessment in the past because of inherent biases. That is why some randomization would be required, which was added to the protocol amendment.
WHY WAS THIS ACCEPTED? As you can see, in the beginning both the FDA and EMA were not ready to approve these products even under an EUA or the Conditional Marketing Authorization of the EMA. What changed? Were they threatened? What were they promised? Is there a spell on the world?
Extra info
https://www.fda.gov/files/drugs/published/Comparability-Protocols-for-Human-Drugs-and-Biologics--Chemistry--Manufacturing--and-Controls-Information-Guidance-for-Industry.pdf
Disturbing and very frightening, who can we trust? So many damaged people from this experimental and badly manufactured injection, my poor friend being one.
In the US we have concluded that the "pandemic" was planned. There were not enough cases to qualify it as a pandemic. Also planned in advance was censorship against anyone who questioned the so-called vaccines. In addition, it was planned well in advance that once the Health and Human Services head declared a "medical emergency," a military plan went into effect. This military plan was not just for logistics as we Americans were told. This was ENTIRELY a military program, under the Joint Chiefs of Staff, with the head of HHS consulting only. The so-called vaccine had been studied for some time under the US bioweapons program. It was clearly a biochemical weapon.
In the US according to legislation, a military countermeasure or medical countermeasure, is no different from a tank or a rifle or a bomb. There is absolutely no pharmaceutical regulatory authority over military countermeasures or medical countermeasures. The military does not need to study the safety of weapons. Thus the gene therapy they labeled as a vaccine was a poison, never intended to be regulated by the FDA. The FDA in fact did no studies to evaluate safety or efficacy, but gave it "Emergency Use Authorization" which made Americans THINK it was a regulated substance. Imagine if the government said, "Take this vaccine even though no one studied it for safety or efficacy?" So they hid behind the EUA. This was fraud in an effort to perpetrate crimes against humanity with the goal of global depopulation. This is an agenda considered extremely important to all those who run the UN and WEF, and WHO. The WHO used to provide programs for the elderly. Now the WHO is planning to provide death for the elderly.
All the health organizations of the world have been turned from life giving to death giving.
You ask "Whom can we trust?" I answer that anyone who purports to present the government's perspective or the EU's perspective, or the perspective of the government of the US regarding "vaccines" or "pandemics" cannot be trusted.
In the US people who are aware of what has been done now are afraid to ever go into the hospital. We know death protocols were used on COVID positive patients that killed them, all these deaths were completely unnecessary.
We also know that the PCR test was a total fraud. It was only used to create a PSYOPS: A psychological operation to fool millions of people around the world that there was a deadly virus killing untold numbers of people.
The PCR test tells you nothing. Never again fall for that.
COVID was a respiratory virus not much different from the cold or flu. There was NEVER A NEED FOR A VACCINE IN THE FIRST PLACE. COVID didn't kill vast numbers of people. The death protocols killed people and the refusal of governments to allow doctors to do their jobs killed hundreds of thousands of people unnecessarily.
Remember this: there is no such thing as a virus that is simultaneously extremely lethal and extremely transmissible. If it is extremely lethal, it is very difficult to transmit. If it is extremely transmissible like the cold virus, it is very NONLETHAL. If a virus could be both extremely lethal and extremely transmissible the human race would have been gone by now. Don't be fooled. There are no such thing as pandemics. This is an excuse for the WHO to become the world's supranational government. This is an excuse to depopulate the globe with poisons and make people like Bill Gates more wealthy. He should be found guilty of crimes against humanity.
Every doctor and public health official who pushed these poisons should be brought to a global tribunal and found guilty of crimes against humanity.