

How is 'Quality and Purity' Measured?
The process is the product

Just before the ‘pandemic’ hit in 2020, I was working as a pharmacy consultant for a smaller hospital pharmacy who had been ignored and poorly managed for many years. One task I had was to help establish policy and procedures for sterile compounding including hazardous (chemotherapy) compounding in a new clean room. The manufacturing we do in hospital is analogous to fill and finish type manufacturing by Pharma but on a smaller scale.
I learned the hard way, how difficult it is to keep the clean room clean, and to ensure all staff were following established protocols and that we were following compendial standards (more about that later).
One day, while the incompetent nurse manager was on vacation and I was placed in charge (sorry nurses should not be in charge of pharmacies), the clean room technician wiped the inside of some plastic bins in the clean room and then showed me this:

Needless to say, there was a whole investigation, and the clean room was closed. I pulled all the epidural which required overtime of the technicians. The administration in the hospital were not happy with me, but could do nothing. Thankfully the chemo hood itself was sterile but the clean room had not been cleaned in at least 3 months, or longer. Furthermore, there was construction above the clean room which increases risk of Aspergillosis (which was found BTW). The staff were no following established protocols, monitoring was lax, and compendial standards were lacking. This is how a drug product can get contaminated.
Contamination, impurities, adulteration, etc
I thought we should review many of these terms because many commentators get this wrong and it is important to be precise in these terms.
Impurities of the Drug Substance (aka the modRNA)
Impurities are EXPECTED components in the process of making a drug or therapeutic.
Process-Related and Product-Related Impurities: are defined as impurities that originate from the manufacturing process and may be derived from reagents used in the in-vitro transcription and purification processes. What would this include? So for the production of the modRNA ONLY we have:
Residual DNA Template: Residual DNA template is a process-related impurity derived from the linearized DNA template added to the in-vitro transcription reaction. Residual DNA template is further controlled through routine testing using qPCR
dsRNA Double stranded RNA is a product-related impurity derived from the in-vitro transcription reaction. Double stranded RNA is further controlled through routine testing using immunoblot analytical procedure
Truncated RNA/Fragmented species: this impurity was added by the regulators but not first identified as an impurities by Pfizer/BioNTech. RNA integrity is measured by capillary gel electrophoresis and agarose gels
Small molecules such as buffers, magnesium acetate, calcium chloride etc
Enzymes used such as T7 polymerase, proteinase K, pyrophosphates
Nucleoside triphosphate (NTPs)/Capping structure such as the N-1-methylpseudouridine
Impurities due to 4, 5 and 6 were not considered sufficiently high enough to be controlled by testing. The ultra/diafiltration are very efficient at removing these elements. In addition a risk assessment of the amounts BEFORE filtration and the theoretical worst case dose was assumed and compared to the safety concern threshold by Pfizer which was accepted by the EMA as non-toxic and did not require continued testing. Whether this assumption is valid is an open question, but these products, especially the small molecules are well known and characterized. However, very small amounts of the enzymes or NTPs could be found in the LNPs and whether that would cause issues intracellularly remains to be seen.
Residual DNA, dsRNA and fragmented RNA were identified by Pfizer/BioNTech and the regulators as impurities which must be controlled and measured. More about that later.
Contamination
Contaminants are defined as any adventitiously introduced materials (e.g., chemical,biochemical, or microbial species) NOT intended to be part of the manufacturing process of the drug substance or drug product. The potential contaminants that may be present in BNT162b2 drug substance (modRNA) are endotoxin and bioburden. These included:
Endotoxin: Endotoxin, from the E coli cell wall, is controlled by monitoring levels before and after established processing steps to ensure no increase in endotoxin levels resulting from various processes involved in the creation and packaging of the drug/therapeutic. This is measured using the limulus amebocyte lysate (LAL) test
Bioburden: defined as the number of bacteria living on a surface that has not been sterilized. The term is most often used in the context of bioburden testing, also known as microbial limit testing, measured by the number of colony forming units (CFUs) which should be 0.
Adulteration
This I defined in my previous substack
Adulteration of the Pfizer/BioNTech vaccine
Critical Quality Attributes for the Drug Substance (aka modRNA)
Now that everyone has an idea of the kinds of impurities and contamination possible in the production of the modRNA, we need to define the Critical Quality Attributes (CQA).
A CQA is a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.
CQAs represent quality AND purity. So a list of these CQAs are proposed by the manufacturer and commented upon by the regulators. Inherent in defining the CQA is
the analytical method used to measure the attribute
the accepted limit, range or distribution (i.e plus or minus of the accepted value)
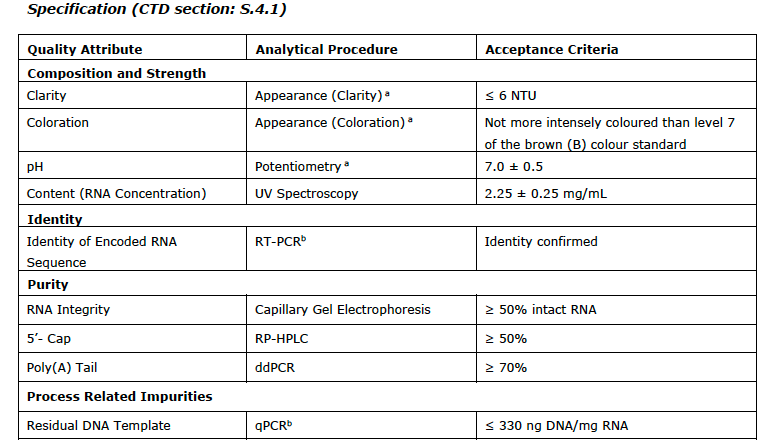
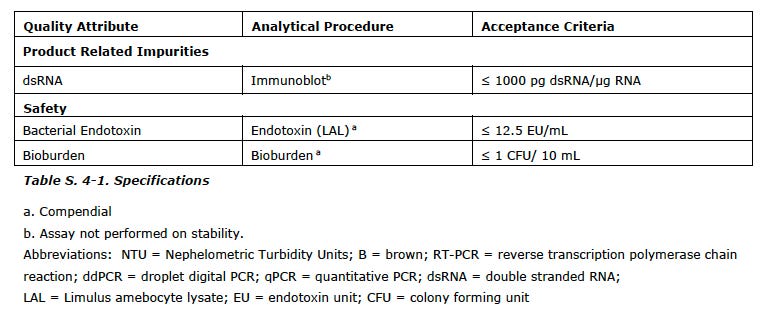
I append the list of the CQAs with the analytical test used and the limits/values from the EMA report at the end of this post.
You can see the range for pH is a full point (!) and modRNA concentration it is plus/minus 11%. You will also note a subscript (a) which means COMPENDIAL.
So how do they decide what is the analytical method to be used, and what is the accepted range or distribution? Well, that is the REAL question regarding the drug substance and drug product. I will expand on that in future substacks.
Also, keep in mind these CQAs are only for the drug substance and not for the final drug product which has ANOTHER slew of impurities and contaminants associated with the lipids plus the final formulation.
Implications
there are a lot of CQAs that are not verified or studied in depth. For a drug product to be given to millions, what are the important variables to control for purity and quality? Should the NTPs be monitored, for example?
Who decides on what the most appropriate analytical method that should be used? There are some compendial standards involved but many are brand new, as this is a new platform. Usually you have a few years to refine analytical methods as they change over time as knowledge of the product improves but this didn’t happen of course. So much for “we have studied mRNA for 20 years+.”
Who decides what the ranges for the CQAs should be? How pure? What quality?
For instance, controlling and measuring residual DNA is required for all biologics, but for this new genetic product, is the analytical method appropriate? Are the acceptance criteria appropriate?
These issues are at the heart of the pDNA impurity debate.
sounds like a witches cauldron is cleaner LOL!