
Why is the manufacturing process of these jabs so important?
Because the process is the product

I have been focussed on the manufacturing of the product, the design of the mRNA, the lipids and the formulation of these pro-vaccines for a very important reason.
THE PROCESS IS THE PRODUCT
What do I mean by that?
Comparing biologics, and the jabs with drugs
Ordinary drugs are small molecules than can be described chemically and very accurately. It really doesn’t matter HOW you synthesize the molecule as long as the chemical structure is the defined drug. Biologics, including vaccines, therapeutic proteins, blood products and the genetic jabs are much more complex.
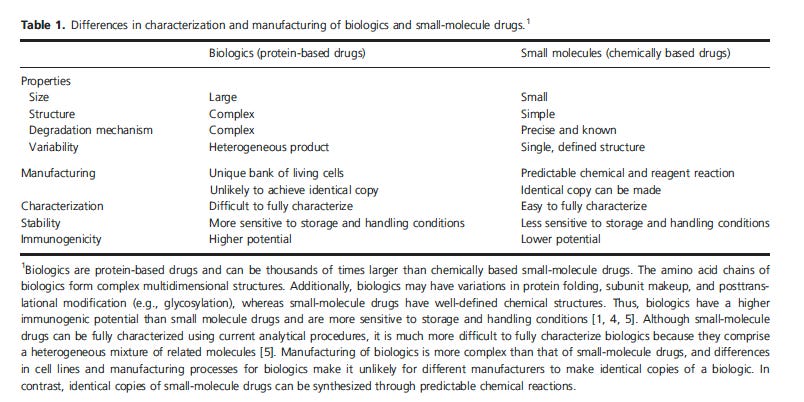
All of the above characteristics apply to the jabs as well.
A biologic drug is manufactured in a living system such as in E coli, or plant or animal cells. Biologics are LARGE, complex molecules or mixtures of molecules.
The average size of a drug is 500 daltons (0.5kDa, the average size of biologic or antibody like adalimumab (Humira) is 148 kDA, and the modRNA is approx 1440kDa but the LNPs themselves are much bigger than that. Plus each LNP has 2-3mRNAs in them. S0 10,000kDa??? (anyone?)
A drug is typically manufactured through chemical synthesis, which means that it is made by combining specific chemical ingredients in an ordered process.
Drugs generally have well-defined chemical structures, and a finished drug can usually be analyzed to determine all its various components. That is the entire structure, including chirality (left or right handedness) etc
By contrast it is difficult, and sometimes impossible, to characterize a complex biologic by testing methods available in the laboratory, and some of the components of a finished biologic may be unknown.
remember that the final product, the spike protein, is made in the body.
how can we measure that each and every patient makes exactly the same Wuhan spike protein? This is not a problem with small molecules because each and every person gets the same chemical.
Therefore, for biologics, "the product is the process." Because the finished product cannot be fully characterized in the laboratory, manufacturers must ensure product consistency, quality, and purity by ensuring that the manufacturing process remains substantially the same over time.
this means Moderna’s product is not the same as Pfizers because some of the starting materials may not be the same, such as enzymes used in the in vitro process
the ONLY way you can ensure the same structure of biological molecule, whether it is a protein, OR the mRNA is ensuring you make it exactly the same way every time.
A biologic has over 250-300 steps that must be controlled. That means the entire step is written out, every ingredient as pure as possible and the step performed exactly the same way every time.
By contrast, a drug manufacturer can change the manufacturing process extensively and analyze the finished product to establish that it is the same as before the manufacturing change. Usually about 40-50 steps.
The living systems used to produce biologics can be sensitive to very minor changes in the manufacturing process.
Small process differences can significantly affect the nature of the finished biologic and, most importantly, the way it functions in the body.
how small? well a small decrease in magnesium ions means a whole lot of DNA in Pfizers vaccines. Any change in LNP size, morphology or amount of pegylated lipid, or purity of the lipids used. Mixing speed in the mixing vat. ANYTHING.
To ensure that a manufacturing process remains the same over time, biologics manufacturers must tightly control the source and nature of starting materials, and consistently employ hundreds of process controls that assure predictable manufacturing outcomes.
Process controls for biologics are established separately for each unique manufacturing process/product, and are not applicable to a manufacturing process/product created by another manufacturer.
What are the important ramifications of the manufacturing process with biologics?
This table is hard to see and describes Remicade (infliximab). This is a therapeutic protein used to treat rheumatoid arthritis and other conditions. The table is hard to see so I will summarize on the issues that also apply to the mRNA jabs.
sequence of the antibody
misfolding or truncation can lead to lower efficacy (we have truncated and fragmented mRNA in the jabs which likely leads to decreased efficacy and possibly toxicity as well)
misfolding can lead to auto-antibodies (effect of codon optimization)
deamination/oxidation (breakdown of the protein) leads to lower efficacy
glycosylation patterns (attachment of sugar molecules to the protein)
can affect toxicity
can elicit an immunological response
Note: we have no idea how much glycosylation occurs on the spike protein made by the modRNA
process impurities
DNA, dsRNA, endotoxin
also solvents, enzymes, substrates etc
biological characterization
ability to elicit neutralization antibodies for the mRNA vaccines
result of the in vivo translation into the authentic Wuhan spike protein for the mRNA vaccines

Quality by Design
Here is a concept in drug development called Quality by Design. This is something Mike Yeadon touches upon and the principles have been ingrained into us making chemo drugs in our clean rooms in the hospital. (note CQA=critical quality attributes)

Based on this standard approach, this is why the presence of the SV40 is so earth-shattering. If it was a risk for quality, then it shouldn’t have been used in the first place.
Implications for Process 1, Process 2, and Process 3: are they NEW drugs?
In essence, the manufacturing process differed substantially between all these processes. And not just the PCR generated DNA to plasmid DNA or PBS buffer to Tris buffer. Changes in the starting materials, purity standards, changes in the supply of vendors for reagents and enzymes. Everything.
If changes in the manufacturing process leads to different products, then are these new products comparable to the previous ones?
For Process 1 vs Process 2, it failed the analytical and physiocochemical comparison
the differences in the amount of impurities of mRNA was substantial
differences in manufacturing of the lipids themselves
freezing of the product and particles
requirement for clinical verification in humans was required but never completed
For what I call Process 3, this was never compared clinically in patients
the differences in available dosage of the mRNA
differences in particle size and risk of toxicity?
potential differences in biodistribution due to differences in size and stability?
did it need to be compared clinically?
Moderna had some similar issues, especially with changes in their lipid manufacturing. They may have been close enough to the original product used in the clinical trials. But they changed the 3’UTR for the bivalents and NEVER got called out on that.
For ALL biologics, and that includes regular vaccines, the manufacturing process defines the product.
I welcome you comments and suggestions.
Pray the rosary.
If you buy a Rolls Royce and Del Boys' Robin Reliant arrives I think that is breach of contract? Obviously not in the pharmaceutical industry then?
Very clear article for us laymen thanks!